Angiologie > Erkrankungen der Arterien > Angeborene Erkrankungen > Teleangiektasien und Teleangiektasiesyndrome
Morbus Rendu-Osler
|
| ||||||||||||
|
| Angiologie > Erkrankungen der Arterien > Angeborene Erkrankungen > Teleangiektasien und Teleangiektasiesyndrome |
|||||||||||
|
Morbus Rendu-Osler
|
||||||||||||
|
| Einleitung | ||
Synonym |
Morbus Osler |
|
 |
||
|
Morbus Osler-Rendu-Weber |
||
|
||
|
ORW-Erkrankung |
||
|
||
|
hereditäre hämorrhagische Teleangiektasien, Typ 1 |
||
|
||
|
hereditäre hämorrhagische Teleangiektasien, Typ 2 |
||
|
||
|
HHT1 |
||
|
||
|
HHT2 |
||
|
||
Englisch |
Osler Weber Rendu disease |
|
|
||
ICD10 |
I78.0 |
|
|
||
Definition |
seltene, autosomal-dominant vererbte Erkrankung mit erhöhter Blutungsneigung und Hämorrhagien aufgrund von Teleangiektasien |
|
|
||
Klassifikationen |
|
|
|
||
| ||
| Epidemiologie | ||
Inzidenz |
|
|
|
||
Prävalenz |
seltene Erkrankung |
|
|
||
Alter |
Manifestation manchmal im Kindesalter (Blutungen), häufiger in der Pubertät / Erwachsenenalter; ab 50 Jahre v.a. als GIT-Blutung |
|
|
||
Häufigkeitsgipfel |
|
|
|
||
Geschlecht |
M = F |
|
|
||
Ethnologie |
keine ethnologische Bevorzugung |
|
|
||
| ||
| Pathologie | ||
Ätiologie |
|
|
|
||
Risikofaktoren |
Blutgruppe 0; im Sommer oft ausgeprägtere Blutungen als im Winter (dixit cand. med. T. Schäfer) |
|
|
||
Vererbung |
AD, Homozygotie wahrscheinlich letal; unterschiedliche Penetranz von minimalen Läsionen ohne Blutungsepisoden |
|
|
||
OMIM |
||
|
||
Chromosom |
9q34.1 |
|
|
||
Pathogenese |
Pathogenese unklar, zwei mögliche Gene:
Beide Gene kodieren für Proteine, die als Typ III bzw. Typ I-Rezeptor des Transforming growth factor β (TGF-β) fungieren und ausschließlich auf vaskulären Endothelzellen exprimiert werden. Die Bindung von TGF-β an den Typ II TGF-β-Rezeptor ist bei der Anwesenheit von Endoglin beschleunigt und führt zu einer Phosphorylierung des Typ I TGF-β-Rezeptors, der activin receptorlike kinase Typ 5 (ALK-5) und ALK-1. |
|
|
||
 siehe auch beiliegender Vortrag vom 23.06.2004 |
||
|
||
Makroskopie |
|
|
|
||
Mikroskopie |
Hautbiopsie manchmal zur Bestätigung der Diagnose hilfreich: dilatierte Kapillaren und Blutgefäßneubildung in der oberflächlichen Kutis; in der Dermis verdickte Gefäßwand der dilatierten Gefäße |
|
|
||
| ||
| Diagnostik und Workup | ||
Kriterien |
Curacao-Kriterien:
Die Diagnose des HHT ist definitiv mit 3 Kriterien, möglich mit 2 Kriterien und unwahrscheinlich mit weniger als 2 Kriterien |
|
|
||
Diagnostik |
Screening auf PAVM (funktional oder Bildgebung) sowie CAVM (MRT) empfohlen |
|
|
||
Körperliche Untersuchung |
|
|
|
||
Bildgebung |
Röntgen-Thorax, Angiographie, CT und/oder MRT |
|
|
||
Blut |
oft mikrozytäre hypochrome Eisenmangelanämie, Thrombozytopenie |
|
|
||
|
erhöhte Transaminasen möglich |
||
|
||
|
ev. unbestimmte Koagulationsstörung und erhöhte fibrinolytische Aktivität (Bedeutung für Blutungen unklar) |
||
|
||
Urin |
Hämaturie möglich |
|
|
||
Stuhl |
Blutiger Stuhl möglich |
|
|
||
Weitere Diagnostik |
|
|
|
||
Nachsorge |
|
|
|
||
Meldepflicht |
|
|
|
||
Pränataldiagnostik |
|
|
|
||
| ||
| Symptome und Befunde | ||
1. |
95% wiederholte Epistaxis (Nasenbluten) |
|
|
||
2. |
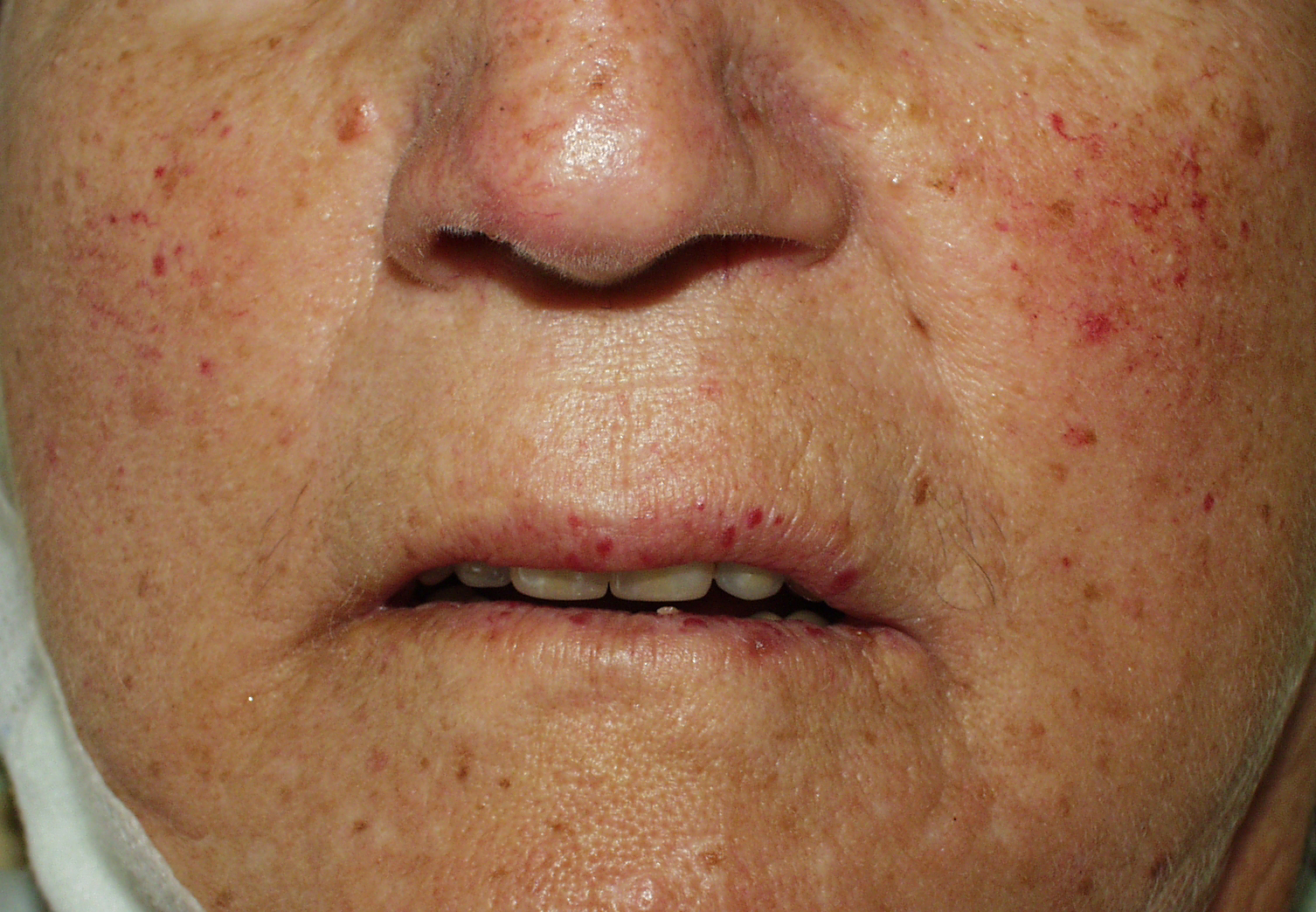 Klicken Sie auf das Bild, um es zu vergrößern. Telangiektasien:
|
|
|
||
3. |
Schleimhäute: immer beteiligt, variabler Befall von gastrointestinalen, respiratorischen und urogenitalen Schleimhäuten; einschließlich Nasenseptum, Mundhöhle und Nasopharynx (Nasenbluten) |
|
|
||
4. |
Pulmonale arteriovenöse Anastomosen (PAVM) mit Fistelbildung: Dyspnoe, Zyanose; 3 Folgen:
|
|
|
||
5. |
Leber (HAVM): Hepatomegalie und Leberzirrhose möglich, ebenso Splenomegalie |
|
|
||
6. |
ZNS: arteriovenöse Malformationen (CAVM) mit Parästhesien, Epilepsien, Steal-Effekt auf Umgebung möglich |
|
|
||
7. |
Schwangerschaft bei Patienten mit pulmonalen arteriovenösen Malformationen: Pulmonale Hämorrhagien mit möglicherweise letalem Ausgang für Mutter und/oder Fetus |
|
|
||
| ||
| Verlauf und Prognose | ||
Stadien |
|
|
|
||
Verlauf |
ca. 1/3 milder, 1/3 mäßiger und 1/3 schwerer Verlauf |
|
|
||
Komplikationen |
|
|
|
||
Prognose |
meist gute Prognose, abhängig vom Grad der systemischen Beteiligung (insbesondere Lunge, Leber, ZNS) |
|
|
||
Prophylaxe |
|
|
|
||
| ||
| Differentialdiagnosen | ||
1. |
Angiokeratoma corporis diffusum (Fabry-Syndrom) |
|
|
||
2. | ||
|
||
3. |
Spider-Nävi bei chronischem Alkoholkonsum |
|
|
||
4. |
CREST-Syndrom: Calcinosis Cutis, Raynaud-Syndrom, Ösophagusmotilitätsstörungen, Sklerodaktylie und Teleangiektasien |
|
|
||
| ||
| Therapien | ||
|
bei leichten Formen keine Therapie notwendig |
||
|
||
Hautveränderungen |
Kautheterisierung, Laserchirurgie (Nd-Laser) |
|
|
||
Epistaxis |
ggf. Nasenseptum-Hauttransplantation aus dem unteren Stamm |
|
|
||
Lunge |
ggf. Silikonballontamponade, Embolisation (ohne Metall-Coil) |
|
|
||
Allgemein |
1. |
bei schwersten Formen ev. Östrogentherapie, prothrombosierender Effekt (1 randomisierte Studie), ggf. andere thrombogene Substanzen |
|
||
2. |
symptomatische Therapie der Anämie mit Eisengabe und ggf. Bluttransfusionen |
|
|
||
3. |
Restriktion der Aktivität bei Blutungen und schwerer Anämie |
|
|
||
4. |
bei PAVM Antibiose vor bakteriämischen Eingriffen empfohlen |
|
|
||
| ||
| Referenzen | ||
Lehrbuch |
||
|
||
Reviews |
||
|
||
|
||
Studien |
||
|
||
|
||
Links |
||
|
||
Adressen |
||
| ||
| Editorial | ||
Autor |
||
|
||
Erstellt |
18.06.2004 |
|
|
||
Reviewer |
Tobias Schäfer (Editor) |
|
|
||
|
Wibke Janzarik, 24.06.2004 |
||
|
||
Linker |
||
|
||
Status |
PRELIMINARY |
|
|
||
Licence |
||
| ||
| Kommentare | ||
| Wibke Janzarik schrieb am 24.06.2004 um 08:43 Uhr: | ||
Review abgeschlossen | ||
artikel nett, insb. foto ;-) | ||
|
||
| Tobias Schäfer schrieb am 24.06.2004 um 10:08 Uhr: | ||
Östrogentherapie | ||
prothrombogene Wirkung, Artikel ergänzt. Vortrag ist angefügt, muss aber noch programmiert werden. | ||
| ||
| |||||||||||||||