Hämatologie > Erkrankungen der Lymphozyten > Non-Hodgkin-Lymphome
Chronische lymphatische Leukämie (CLL)
|
| ||||||||||||
|
| Hämatologie > Erkrankungen der Lymphozyten > Non-Hodgkin-Lymphome |
|||||||||||
|
Chronische lymphatische Leukämie (CLL)
|
||||||||||||
|
| Einleitung | ||
Synonym |
Chron. Lymphadenose |
|
 |
||
Englisch |
|
|
|
||
ICD10 |
C91.1 |
|
|
||
Definition |
Meist leukämisch verlaufendes B-Zell-Lymphom von niedrigem Malignitätsgrad mit klonaler Proliferation und Akkumulation immuninkompetenter B-Lymphozyten in Lymphknoten, Milz und Knochenmark. |
|
|
||
Klassifikationen |
|
|
|
||
| ||
| Epidemiologie | ||
Inzidenz |
Zunehmende Inzidenz im höheren Lebensalter: Im 5. Lebensjahrzehnt ca. 5/ 100 000 jährlich, im 8. Lebensjahrzehnt ca. 30/ 100 000 jährlich. |
|
|
||
Prävalenz |
|
|
|
||
Alter |
|
|
|
||
Häufigkeitsgipfel |
|
|
|
||
Geschlecht |
M : F = 2: 1 |
|
|
||
Ethnologie |
In Japan sehr selten |
|
|
||
Sonstiges |
Häufigste Leukämieform |
|
|
||
| ||
| Pathologie | ||
Ätiologie |
unklar |
|
|
||
Risikofaktoren |
|
|
|
||
Vererbung |
|
|
|
||
Chromosom |
Ca. 40% der Patienten haben chromosomale Veränderungen, z.B. Trisomie 21, Translokationen: t(11;14), t(14;19) |
|
|
||
Pathogenese |
|
|
|
||
Makroskopie |
|
|
|
||
Mikroskopie |
In der Leberhistologie: periportale Lymphozyteninfiltration (bei der CML diffuse Infiltration) |
|
|
||
Sonstige Klassifizierung |
Stadieneinteilung nach Binet (1981):
Durch zusätzliche Parameter kann das Binet-Stadium A unterteilt werden:
|
|
|
||
| ||
| Diagnostik und Workup | ||
Kriterien |
Klinik und BB (anhaltende Lymphozytose) |
|
|
||
Diagnostik |
In 70% symptomloser Zufallsbefund bei Diagnosestellung. Lymphknotenschwellungen sind im Verlauf einer CLL immer vorhanden. Serumeiweissveränderungen (B-Zelldefekt!) in 50%: Beta2-Mikroglobulin-Serumspiegel, Thymidinkinase und der CD23-Serumspiegel korrelieren bei norm. Nierenfunktion mit der Gesamttumormasse. |
|
|
||
Körperliche Untersuchung |
Lymphknotenschwellungen (derb, indolent): initial 50%, später alle Patienten |
|
|
||
Bildgebung |
Im Röntgen-Thorax/ CT: mediastinale LK-schwellungen (25%) |
|
|
||
Blut |
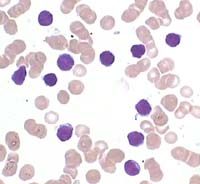 © 2003 A Med World AG. Verwendet mit freundlicher Genehmigung. Permanente Leukozytose mit einem hohen Lymphozytenanteil (meist 70 - 90%) und permanente Erhoehung der absoluten Leukozytenzahl >10 000/ Mikroliter. Typisch (nicht obligat) sind Gumprecht'sche Kernschatten (gequetschte Lymphozytenkerne). |
|
|
||
Weitere Diagnostik |
Hohe diagn. Aussage: Knochenmarkzytologie/-histologie: Anteil reifer Lymphozyten >/= 40% aller kernhaltigen Zellen bei norm. oder erhöhtem Zellgehalt. Noduläre Ausbreitung der CLL im KM ist prognostisch günstiger als diffuse Ausbreitung. Durchflusszytometrie mit Immunphänotypisierung der Lymphozyten: Chromosomenanalyse: Ca. 40% der Patienten haben chromosomale Veränderungen, z.B. Trisomie 21, Translokationen: t(11;14), t(14;19) |
|
|
||
Nachsorge |
|
|
|
||
Meldepflicht |
|
|
|
||
Pränataldiagnostik |
|
|
|
||
| ||
| Symptome und Befunde | ||
1. |
evtl. Leistungsminderung, Nachtschweiß |
|
|
||
2. |
Lymphknotenschwellungen (derb, indolent) |
|
|
||
3. |
evtl. Splenomegalie, geringe Lebervergrösserung |
|
|
||
4. |
Hauterscheinungen: Pruritus, chron. Urtikaria, mukokutane Purpura, Herpes zoster, Herpes simplex, Mykosen, Erythrodermien, knotige Hautinfiltrate |
|
|
||
5. |
evtl. Parotisschwellung und Tränendrüsenbefall (Mikulicz-Syndrom) |
|
|
||
| ||
| Verlauf und Prognose | ||
Stadien |
|
|
|
||
Verlauf |
|
|
|
||
Komplikationen |
Noduläre Ausbreitung der CLL im KM ist prognostisch günstiger als diffuse Knochenmarkinfiltration. Mittlere Ueberlebenszeit: Die CLL ist mit Abstand die gutartigste Erkrankung unter allen Leukosen. Ueberlebenszeiten sind sehr variabel und stadienabhängig. Die Hälfte aller Patienten stirbt an Infekten. Unter der klassischen Chemotherapie mit Chlorambucil gibt es keine Heilungen; echte Heilungschancen in seltenen Fällen durch allogene KM-transplantation, bei hoher therapiebedingter Mortalität. |
|
|
||
Prognose |
|
|
|
||
Prophylaxe |
|
|
|
||
| ||
| Differentialdiagnosen | ||
1. |
Reaktive Lymphozytosen (Klinik, meist polyklonale T-Zellmarker) |
|
|
||
2. |
Lymphknotenschwellungen anderer Genese (LK-histologie) |
|
|
||
3. |
CML (typ. BB, Philadelphia-Chromosom) |
|
|
||
4. |
DD eines generalisierten Pruritus:
|
|
|
||
| ||
| Therapien | ||
|
Patienten mit smoldering CLL werden nicht therapiert. Grundsätzlich gilt: die Lymphozytenzahl allein ist kein Therapieindikator. |
||
|
||
medikamentös |
1. |
konventionelle Chemotherapie bei symptomatischen Patienten im Stadium B, alle Patienten im Stadium C:
|
|
||
2. |
Glukokortikoide bei AIHA oder Autoimmunthrombozytopenie; falls erfolglos, hochdosiert Immunglobuline, evtl. Splenektomie. |
|
|
||
Knochenmarkstransplantation |
|
|
|
||
Strahlentherapie |
Bei lokal niedrig dosierte Bestrahlung grosser Lymphome oder einer grossen Milz. |
|
|
||
| ||
| Referenzen | ||
Lehrbuch |
Herold 2002, S. 71ff |
|
|
||
Reviews |
||
|
||
Studien |
||
|
||
Links |
||
|
||
Adressen |
||
| ||
| Editorial | ||
Autor |
||
|
||
Erstellt |
13.11.2002 |
|
|
||
Reviewer |
||
|
||
Linker |
||
|
||
Status |
TRACK3 |
|
|
||
Licence |
||
| ||
| Kommentare | ||
| ||
| |||||||||||||||||||||