Pädiatrie > Hämatologie > Hämatologische Erkrankungen im Kindesalter > Leukämien im Kindesalter
Akute lymphatische Leukämie (ALL)
|
| ||||||||||||
|
| Pädiatrie > Hämatologie > Hämatologische Erkrankungen im Kindesalter > Leukämien im Kindesalter |
|||||||||||
|
Akute lymphatische Leukämie (ALL)
|
||||||||||||
|
| Einleitung | ||||||||||||||||||||||||||||||||||||||||||
Synonym |
|
|||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||
Englisch |
acute lymphoblastic leukemia |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
ICD10 |
C95.9 |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Definition |
Systematisierte diffuse autonome Proliferation von hämatopoetischen, leukozytären Stammzellen mit Ausschwemmung unreifzelliger Blasten ins Blut. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Klassifikationen |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Epidemiologie | ||||||||||||||||||||||||||||||||||||||||||
Inzidenz |
Inzidenz der akuten Leukämien 4/ 100 000/ Jahr, in den USA 2.8/ 100 000/ Jahr |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Prävalenz |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Alter |
Kinder > Erwachsene |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Häufigkeitsgipfel |
2-5 Jahre |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Geschlecht |
Jungen > Mädchen |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Ethnologie |
in den USA Weisse doppelt so häufig betroffen wie Farbige |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Sonstiges |
80% der akuten Leukämien im Kindesalter sind ALL (häufigste maligne Erkrankung im Kindesalter). |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Pathologie | ||||||||||||||||||||||||||||||||||||||||||
Ätiologie |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Erreger |
HTLV1- oder 2-Viren verursachen die T-ALL (C91.5), die in Südjapan und der Karibik endemisch auftritt. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Risikofaktoren |
genetische und immunologische Syndrome (z.B. Trisomie 21, Neurofibromatose I, Bloom Syndrom, Ataxia teleangiectatica) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Vererbung |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Pathogenese |
Neoplastische Transformation der hämatopoetischen Stammzellen und Expansion des malignen Zellklons auf Kosten der normalen Hämatopoese. Progrediente KM-Insuffizienz führt zu klin. Symptomen. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Makroskopie |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Mikroskopie |
Im Blut/ KM finden sich wenig differenzierte oder undifferenzierte Blasten mit grossen atypischen Nukleolen und schmalem, basophilen Zytoplasmasaum. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Sonstige Klassifizierung |
Einteilung nach morphologischen, immunologischen, biochemischen und cytogenetischen Kennzeichen: ALL-Subtypen: |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
a) Einteilung nach der Morphologie (French-American-British System):
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
b) Immuntypisierung der ALL (mit monoklonale Antikörper gegen CD-Marker):
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
c) Cytogenetische Kriterien zur Einteilung in verschiedene Risikogruppen mit unterschiedlichen Behandlungsschemata: Karyotypanalyse, Nachweis von bekannten Chromosomenannomalien mittels FISH und anderen molekularen Techniken. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Geschichte |
Leukämie bedeutet "weisses Blut" und bezieht sich auf die verbreiterte Leukozytenmanschette (buffy coat) auf der Erythrozytensäule nach Zentrifugieren des Blutes bei Leukämiepatienten mit sehr hohen Leukozytenzahlen. Virchow prägte den Begriff bei einer CML. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Diagnostik und Workup | ||||||||||||||||||||||||||||||||||||||||||
Kriterien |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Diagnostik |
Klinik, BB und Knochenmarkbefund mit Zytochemie und Immundiagnostik. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Körperliche Untersuchung |
s. Symptome und Befund: z.B. Symptome einer Anämie (Blässe, Tachykardie, Herzgeräusch), Vergrösserung von Milz oder Leber, Lymphknotenschwellung |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Bildgebung |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Blut |
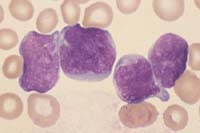 © 2003 A Med World AG. Verwendet mit freundlicher Genehmigung. Blutbild, Knochenmarkzytologie und -histologie:
Zytogenetik: Das TEL-AML 1-Fusionsgen als Folge der Translokation t(12;21) ist die häufigste genetische Aberration bei kindlicher B-ALL (30%). BSG erhöht, ev. Harnsäure und LDH erhöht (vermehrter Zellumsatz). |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Liquor |
Liquorzytologie (! KI bei thrombopenischer Blutungsneigung) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Weitere Diagnostik |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Nachsorge |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Meldepflicht |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Pränataldiagnostik |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Symptome und Befunde | ||||||||||||||||||||||||||||||||||||||||||
Allgemeinsymptome |
1. |
kurze Anamnese! |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Abgeschlagenheit, Fieber, Nachtschweiß |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Verdrängungssymptome |
1. |
Symptome infolge der Verdrängung der normalen Haematopoese: Infektanfälligkeit, Entzündungen an Haut-Schleimhautübergaengen, Pilzinfektionen (Soor durch Candida albicans) |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Symptome durch die Anämie: Blässe, Dyspnoe, Müdigkeit |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
3. |
Symptome infolge der Thrombozytopenie u./o. Verbrauchskoagulopathie: Blutungen |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
weitere |
1. |
Knochenschmerzen in 20-30% durch Beteiligung des Periosts oder durch aseptische Osteonekrose im Rahmen einer akuten Leukämie: Knochenschmerzen sind im Wachstumsalter häufig, können jedoch Symptom einer Leukämie sein. Bei persistierenden Knochenschmerzen und auffälligem Blutbild immer Knochenmarkspunktion. |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Lymphknotenschwellungen in 30-50% (ein LK gilt als vergrößert, wenn er im größten Durchmesser >10mm bzw. >15mm inguinal misst) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
3. |
Kopfschmerzen: selten (<5%); Symptome eines erhöhten Hirndruckes sind außerdem Uebelkeit, Erbrechen, Lethargie, Papillenödem, Nackensteifigkeit. Meningitis leucaemica bes. bei ALL mit leukämischen Infiltraten am Augenhintergrund und neurologischen Symptomen. Evtl. Manifestation mit Hirnnervenausfällen. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
4. |
Splenomegalie, seltener Lebervergrößerung (Kinder > Erw.) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
5. |
unilaterale schmerzlose Hodenvergrößerung: seltenes Erstsymptom (Abklärung mittels Ultraschall und Biopsie). |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
6. |
Mediastinalverbreiterung durch Infiltration von Tumorzellen und Lymphadenopathie. Die Unterscheidung zw. einer ALL und einem Non-Hodgkin-Lymphom kann schwierig sein. Maligne Lymphoblasten in T-Zell-Lymphomen und T-Zell-ALL lassen sich nicht unterscheiden; die Klassifzierung als Leukämiepatient richtet sich nach dem Ausmaß des Knochenmarkbefalls. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
7. |
Leukämische Haut- und Organinfiltrationen |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Die Symptome sind meist unspezifisch (Fieber, Abgeschlagenheit, Blutungen, Lymphadenopathie, Knochenschmerzen, o.ä.). Bei unerklärlicher Persistenz eines dieser Symptome sollte eine Leukämie ausgeschlossen werden. | ||||||||||||||||||||||||||||||||||||||||||
| Verlauf und Prognose | ||||||||||||||||||||||||||||||||||||||||||
Stadien |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Verlauf |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Komplikationen |
Infektionen aufgrund Neutropenie, insb. unter Induktionstherapie. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Blutungen durch Thrombozytopenie (v.a. bei Thrombozytenzahlen <10 000/Mikroliter). |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Tumorlysesyndrom: metabolische Komplikationen durch rasche Tumorzelllyse unter Chemotherapie, wie Hyperphosphatämie, Hypokalzämie (durch Ablagerung von Kalziumphosphat), Hyperurikämie, Hyperkaliämie und akutes Nierenversagen (Uratnephropathie). |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Thrombose (inkl. Sinusvenenthrombose mit Hämorrhagie, tiefe Venenthrombose, Lungenembolie) bei Beginn einer Chemotherapie mit L-Asparaginase, aufgrund Hemmung der Synthese von Plasmaproteinen. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Anaphylaxie auf verschiedene Chemotherapeutika, insb. Asparaginase, Podophylotoxine und Urikase. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Komplikationen nach allogener Knochenmarktransplantation/ Stammzelltransplantation:
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Prognose |
Die Ueberlebensrate von Kindern mit ALL hat sich in den letzten Jahrzehnten dramatisch verbessert. Derzeit beträgt die 5-Jahres-Ueberlebensrate 78-85%. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Nach Chemotherapie im Kindesalter hat der immunolog. Nachweis einzelner residualer Leukämiezellen nach Terapie (MDR: minimal residual disease) prognostische Bedeutung. Patienten mit < 1 Tumorzelle/ 10 000 Lymphozyten haben eine günstige Prognose, Patienten mit >/= 1 Tumorzelle/ 10 000 Lymphozyten haben eine ungünstige Prognose. ungünstige Prognosefaktoren:
t (12;21) ist die häufigste genetische Aberration bei kindlicher B-ALL (30%) und geht mit einer guten Prognose einher. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Spätschäden: Da immer mehr Kinder auch langfristig ihre ALL überleben, kommt es zunehmend auch zur Manifestation von Spätschäden
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Prophylaxe |
Zur Verhinderung eines Tumorlysesyndroms vor Beginn der Chemotherapie Flüssigkeit- und Allopurinolgabe oder rekombinante Uratoxidase (=Urikase !KI bei Glc-6-P-Dehydrogenasemangel, da Hämolyse). |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Differentialdiagnosen | ||||||||||||||||||||||||||||||||||||||||||
1. |
Myelodysplasiesyndrom (bei Panzytopenie), idiop. thrombozytopenische Purpura, aplastische Anämie |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Juvenile Rheumatoidarthritis oder Osteomyelitis bei Knochenschmerzen |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
3. |
Mononukleose: Bei Lymphknotenschwellungen mit atypischen Lymphozyten im BB (Buntes BB mit Reizformen der Lymphozyten, bei meist normalen Thrombozyten und Erythrozyten, pos. Paul Bunnell-Test bzw. AK-Titer gegen EBV |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
4. |
Pertussis, Parapertussis |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
5. |
Akute infektiöse Lymphozytose |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
6. |
Andere maligne Erkrankungen mit Knochenmarkinfiltration (z.B. Neuroblastom, Retinoblastom, Rhabdomyosarkom, Ewing-Sarkom) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
7. | ||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Therapien | ||||||||||||||||||||||||||||||||||||||||||
|
Behandlung in Zentren nach Therapieprotokollen innerhalb von Studiengruppen. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
Wichtig für die Risikostratifikation sind die Leukozytenzahl, das Alter bei Diagnosestellung, die cytogenetische Klassifizierung der ALL, die Immunophenotypisierung und das Ansprechen auf die Induktionstherapie. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
symptomatisch |
1. |
sorgfältige Hygiene, keimarme Räume |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Infektprophylaxe durch selektive Dekontamination von Oropharynx und GI-Trakt mit lokal wirksamen Antibiotika/ Antimykotika |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
3. |
Substitution von Erythrozyten und Thrombozyten nach Bedarf, Gabe von G-CSF und GM-CSF |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
4. |
Bei neutropenischem Fieber: Gabe von Breitbandantibiotika |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
5. |
Prophylaxe einer Uratnephropathie unter zytostatischer Therapie: reichlich Flüssigkeitszufuhr und Gabe von Allopurinol |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Chemotherapie |
Ziel: Erreichen einer kompletten Remission (= Normalisierung von BB/ KM mit < 5% blastären Zellen im KM) |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Chemotherapie bei kindlicher ALL |
1. |
Remissionsinduktionstherapie mit Vincristin, Prednison und L-Asparaginase (oder Daunorubicin) führt in ca. 95% der Fälle zur Vollremission, was durch eine zytostatikainduzierte Knochenmarkaplasie erreicht wird. Prophylaktische Schädelbestrahlung (24 Gy) und intrathekale Injektion von Methotrexat wegen eventueller ZNS-Beteiligung. Da jedoch neuropsychologische Spätschäden gehäuft beobachtet wurden, wird die standardmässige kraniospinale Radiotherapie mittlerweile in vielen Protokollen reduziert oder sogar ganz darauf verzichtet. |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Weiterbehandlung nach Remission: Wiederholungen der Induktionstherapie zur Konsolidierung und Erhaltungstherapie ueber min. 24 Monate, z.B. mit Methotrexat, 6-Mercaptopurin |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Chemotherapie bei Akuter Leukämie im Erwachsenenalter |
davon sind 20% ALL |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
1. |
Remissionsinduktionstherapie: versch. Therapieprotokolle |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
2. |
Remissionserhaltende Therapie: Verschiedene Strategien werden erprobt: kontinuierliche oder diskontinuierliche Behandlung; mit oder ohne intermittierende intensive Zytostatikazyklen |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Allogene Knochenmarks-transplatation/ Stammzell-Transplantation nach myeloablativer Therapie |
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Allogene Stammzelltransplantation nach nicht-myeloablativer Therapie |
Nach einer nicht-myeloablativen Konditionierungstherapie (z.B. mit Fludarabin, Busulfan und ATG) erfolgt die allogene SZT. Erste Ergebnisse deuten auf eine bessere Verträglichkeit hin (als nach myeloablativer SZT), Rezidive können erfolgreich mit "donor lymphozyte infusion" behandelt werden. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Autologe Stammzelltransplantation |
Vorteile der SZT gegenüber KMT:
Vorteile der autologen gegenüber der allogenen Transplantation:
Nachteile der autologen gegenüber der allogenen Transplantation:
|
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Referenzen | ||||||||||||||||||||||||||||||||||||||||||
Lehrbuch |
Herold 2002, S. 74ff. |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Reviews |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Studien |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Links |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Adressen |
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Editorial | ||||||||||||||||||||||||||||||||||||||||||
Autor |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Erstellt |
01.12.2002 |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Reviewer |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Überarbeitet |
Wibke Janzarik, 22.06.2004 |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Linker |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Status |
TRACK3 |
|||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Licence |
||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| Kommentare | ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||